2001年FDA批准伊马替尼标志着激酶ATP竞争性、正构配体作为药物的有效性。这一首次成功引发了对激酶抑制多个方面的研究,包括了解正构点结合的微妙性和替代作用机制(MOA),如假激酶、别构和共价结合,以及抑制蛋白质-蛋白质相互作用。目前,超过70种小分子蛋白和脂质激酶抑制剂已被批准用于临床(图1)。虽然三种机制(正构、变构和共价)在获批药物中都有体现,但变构作用机制在获批药物数量和靶点多样性方面都相对不足,其中仅包括2013年开始批准的四种III型MEK1/2抑制剂(trametinib、cobimetinib、binimetinib和selumetinib),以及2021年10月批准的一种IV型BCR-ABL1抑制剂(asciminib)。预计TYK2假性激酶抑制剂deucravacitinib将于2022年提交审批。

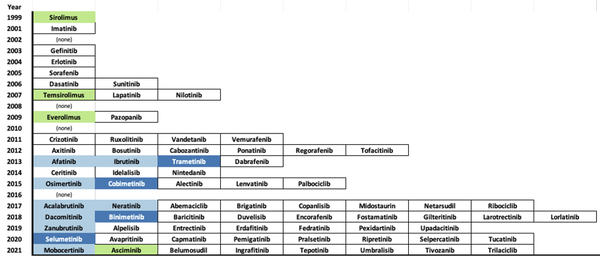
图1. 到2021年10月29日,FDA批准的小分子蛋白和脂质激酶抑制剂,按首次批准的年份排列。III型抑制剂显示为深蓝色,IV型抑制剂显示为浅绿色,共价抑制剂显示为浅蓝色,I型和II型抑制剂为白色背景(图片来源:J. Med. Chem.)。
已获批准的激酶药物和因变构结合而产生的有效目标的代表性不足,这可能是变构抑制剂发现所面临的各种挑战的结果。对X射线共晶体结构中的变构抑制剂的分析突出了蛋白质构象重排的重要性和预测变构位点的挑战。因此,筛选策略必须考虑到蛋白质的动态性质和与结合到有时诱导的口袋相关的动力学。识别在细胞实验中表现出功能活性的非正交配体需要大量的努力,从确定筛选哪些化合物集合和开发适当的筛选范式,到确认结合、生化和细胞实验之间的体外流动方案连接。尽管如此,变构配体可以在激酶的选择性概况中产生好处,并在体内环境中产生相关的药理学和耐受性。
本文回顾了激酶配体结合的类别,探讨了激酶的变构位点,介绍了临床上取得进展并获得FDA批准的变构配体相关的历史,揭示了识别和验证此类配体的挑战。此外,还回顾了识别异体配体的筛选方法,以及为药物发现建立一个强大的、连接的流程方案所需的一系列工具。

2.1 激酶配体结合口袋的类型
"ATP口袋 "和 "正构位点 "通常可以互换使用。正构位点是指内源性配体结合的受体部位,对于激酶来说,内源性配体是ATP。激酶正构配体结合的分类逐渐演变为反映蛋白质构象的细微差别,以及描述变构配体的结合。最熟悉的正构配体的类别是I型和II型。虽然两者都是与ATP竞争,但I型配体与酶的活性形式结合。激酶的 "活性构象"是α螺旋定位在催化赖氨酸和谷氨酸之间形成盐桥。
II型配体与酶的非活性形式结合;激酶是非活性的,因为含有Asp-Phe-Gly(DFG)的柔性的N端环的方向是离开或远离活性部位。这样一来,ATP口袋可以部分地被环路阻断,同时创造了一个新的配体结合口袋。还有更多的I型和II型的亚类,描述了正构部位构象的其他细微差别。目前FDA批准的蛋白和脂质激酶抑制剂中约有80%被归为I型或II型。
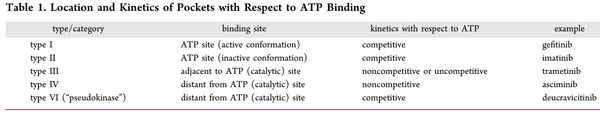
在ATP竞争机制方面还有其他的重大变化,表1中提供了一些例子。包括JAK异构体和TYK2在内的少数几种激酶拥有一个非催化性的ATP结合口袋,或一个假激酶口袋。百时美施贵宝(BMS)已经用BMS-986165(deucravicitinib)证明,结合到TYK2的JAK同源结构域2(JH2结构域)可以有效地调节该蛋白的功能活性,并转化为体内的疾病调节作用。尽管这个部位是ATP竞争性的,因为它不是催化性的,它也可以被认为是变构的,这样的配体被归类为VI型抑制剂。V型抑制剂是二价的,横跨激酶的两个区域,包括正构位点。虽然V型体外工具化合物已经发表,但目前还没有一个进入临床开发。
表1. 袋的位置和动力学与ATP结合的关系(来源:J. Med. Chem.)
与正构部位结合剂相反,变构调节是通过配体在酶的活性部位以外的部位结合而发生的。因此,该部位可以位于蛋白质的任何地方,被描述为III型或IV型。III型配体结合在ATP位点的近端或紧邻的位点。X射线晶体学显示,配体可以以DFG-in(活性)或DFG-out(非活性)的构象结合到III型口袋中(参见下文)。这两种构象的共同点是,结合可以强制改变α-C-螺旋的位置,使其采用非活性构象,阻止对催化作用至关重要的谷氨酸在ATP位点上正确排列。III型抑制剂倾向于表现出ATP非竞争性动力学,结合不能有效利用ATP的酶的形式(见下面关于构象的讨论)或酶-底物复合物。也有不太常见的情况,动力学分析显示配体是ATP非竞争性的,即配体只结合酶-底物复合物。然而,仅靠动力学分析不足以确认III型结合,还需要更多的生物物理学特性分析。FDA批准的MEK1/2抑制剂trametinib、cobimetinib、binimetinib和selumetinib是III型配体的明显例子。
IV型配体在远离ATP位点的地方结合。虽然mTOR抑制剂sirolimus、everolimus和temsirolimus被归类为IV型抑制剂,但应该注意的是,它们首先与FKBP12结合,然后与mTOR结合,导致对催化作用的抑制;它们没有结合到mTOR本身的口袋。Asciminib代表一种真正的IV型配体,它结合在激酶BCR-ABL1的一个部位,该部位远离正位口袋,并通过一个残基网络调节催化活性。III型和"真正的"IV型配体都被认为是通过调节残基网络通过蛋白质向活性部位的移动来破坏催化作用。这种对残基运动的影响也被描述为 "变构网"。图2示意性地描述了一个典型的激酶--BCR-ABL1上的结合口袋的位置。
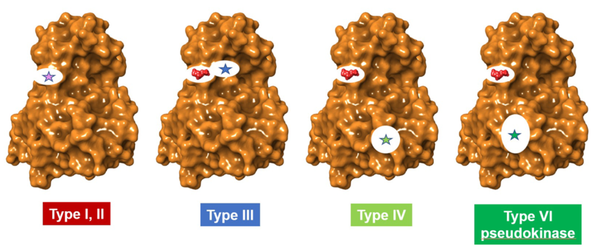
图2. 相对于正构部位的变构口袋的位置。对于I型和II型配体,正构口袋用星星表示。对于III、IV和VI型,ATP显示在红色的空间填充模型中,而变构位点则用星形表示。VI型口袋结合ATP,但不直接促进蛋白质底物的磷酸化。(图片来源:J. Med. Chem.)
2.2 变构调控的理论
一些评论总结了变构调节理论的演变。20世纪60年代提出的最初概念认为,蛋白质以有限的离散构象存在,其中一个构象能够适应具有互补形状的配体的结合。蛋白质在构象间的运动被认为是 "协同的"(Monod-Wyman-Changeaux模型)或 "顺序的"(Koshland-Nemethy-Filmer模型)。这些模型发展到 "群体转移 "模型,其中与构象状态相关的能量景观被理解为向配体-蛋白复合物所青睐的构象重新分布。这个模型中隐含的是将蛋白质视为存在于一系列构象中,而不是简单的结构状态。在Tong和Seeliger的分析中,构象状态之间有更大的流动性,它可以被认为是应用于激酶的群体转移思想的一个变种。
虽然III型激酶口袋由于α-C螺旋和催化谷氨酸残基的移动而具有非活性构象,这在配体结合时发生,但上述观察和模型并没有解决为什么占据IV型口袋会导致催化活性降低的问题。最近的一个模型提供了一个潜在的解释,它创造了一个与小提琴相类似的模型。在这个模型中,不仅有构象选择,而且在配体结合后,残基的振动跨越蛋白质到活性部位的改变。这归因于激酶的残基("群体")作为半刚性体移动。Fersht和他的同事报告了一个例子,他们发现当12埃外的天冬氨酸残基被突变为丝氨酸时,水解酶中的一个催化组氨酸残基的pKa变化了0.40个单位(0.55千卡/摩尔),这说明了调节远离感兴趣部位的电荷的影响。因此,异源配体结合的影响可能不仅仅是改变构象群,还可能影响正交部位的电荷。
由此可见,并不是所有的口袋都是平等的,它们影响蛋白质动力学的方式具有功能性后果。例如,口袋可以位于整个蛋白质的表面,但在浅口袋中的配体结合可能不会在基于细胞的试验中表现出功能活性。
2.3变构口袋的普遍性
最近的出版物调查了III型和IV型口袋的普遍性,采取了几种不同的方法。第一种方法是以实验性配体-蛋白质X射线共晶体结构为基础,了解构象的相似性和结合袋的普遍性。第二种是通过与配体EAI001的比较来前瞻性地识别III型口袋,该配体在参考结构5D41中被发现与EFGR T790M的III型口袋结合。其他识别 "热点"或潜在可药用口袋的计算方法也有报道,但总的来说,这些方法并不能确保结合到这种口袋会导致功能活性。
2017年,诺华公司的一个团队研究了文献和蛋白质数据库中的III型和IV型结构。在他们2015年10月收集数据时,发现10个激酶有III型口袋,7个有IV型口袋。人们注意到,III型或 "back pocket"配体与特定的构象以及与催化部位的接近程度有关。III型口袋的特点是α-C-螺旋移位,相对于有正构配体的结构,DFG模块被描述为 "out"。这些DFG-out、变构back pocket配体与II型激酶抑制剂的区别在于ATP位点被闭塞,与催化残基相关的氢键被破坏了。具有III型口袋的十个激酶包括ITK和MEK1/2(DFG-in),以及AKT、FAK、IGF1R、LIMK、PAK1、RIPK1和p38α(DFG-out)。CDK2呈现出一种非典型的情况,其中一份配体,8-苯胺-1-萘磺酸(ANS),在邻近正交位点的口袋中结合得很弱(37μM),并沿着α-C螺旋而不是DFG图案延伸。对于拥有 "典型的"III型口袋的激酶来说,移位的α-C-螺旋为配体的结合创造了空间,并且α-C-螺旋的 "外"构象在ATP位点结合剂存在或不存在的情况下显然是稳定的。这一观察表明,只有有限的激酶有可能容纳必要的α-C-螺旋构象以进行背袋结合。鉴定了数量较少的具有功能特征的IV型结合物(ABL、AMPKα1/2、CHK1、JNK1和PDK1),以及另外两个无功能的口袋(JAK3和PKACα)。图3中的所有激酶家族都至少有一个成员被证明拥有III型或IV型口袋。
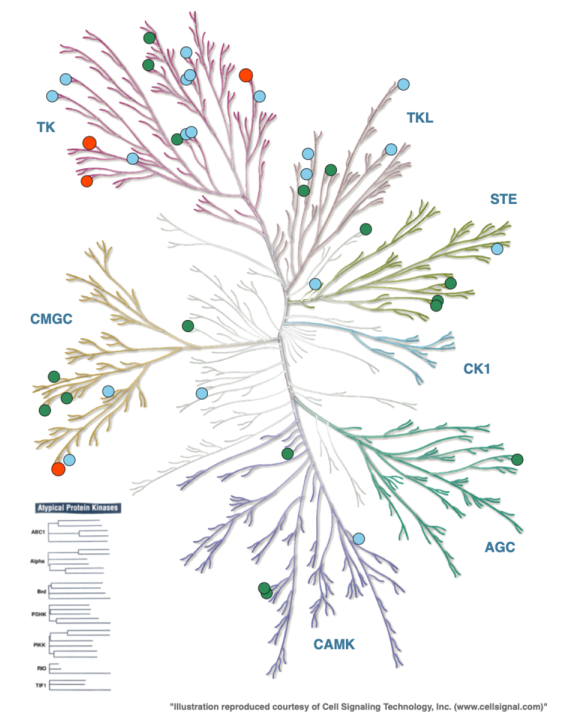
图3. 具有已知和预测的III型位点的激酶。绿色圆圈:具有已知III型位点的激酶,用晶体结构证明;蓝色圆圈:具有预测III型位点的激酶;红色圆圈:同时具有已知和预测III型位点的激酶。用KinMap生成的图形。(图片来源:J. Med. Chem.)
最近,波恩大学的一个小组更新并扩大了这种对激酶实验性X射线共晶体结构的分析,以确定任何类型的变构配体。他们的分析在19个激酶中发现了90个III型(或VI型)配体,以及81个IV型配体在各种激酶的不同位置。在诺华分析之后的几年里, III型口袋的X射线共晶体结构在TrkA、EGFR(T790M)、WNK、ERK5和IRE1中被报道(图3中的绿点)。波恩研究小组注意到发现这些口袋的频率,哪些配体在多个激酶中被发现,以及可能是变构结合的特权结构的核心。许多IV型配体是分子量在200至300达之间的片段。也许不足为奇的是,与这些低分子量配体相关的结合袋可以被描述为浅层,可能与该目标的功能活性无关。
Rastelli小组通过比较EGFR T790 M突变体与III型抑制剂EAI001(PDB 5D41)的X射线共晶体结构,提出了一种识别III型变构口袋的前瞻性方法。利用由一组与EAI001相互作用的24个残基定义的变构位点,他们探测了是否有其他具有类似变构位点构象的激酶结构。然后对那些在ATP结合袋近端区域具有类似构象的残基的激酶晶体结构进行了计算性几何相似性匹配。在应用额外的相似性标准后,确定了20多种激酶,人们可以尝试筛选和确定III型配体(图3中的蓝点)。研究小组还对这些激酶的已知的、假定的正交抑制剂进行了对接研究,以确定是否有任何抑制剂事实上适合假定的III型部位。没有一个抑制剂被发现有纯粹的变构结合模式,尽管少数激酶(EGFR、Src、HCK、BTK和BRAF)有大量的ATP竞争性配体延伸到变构口袋。作者认为,这提供了修改已知配体的机会,以通过创造额外的相互作用和改善活性或选择性来达到III型口袋。没有对这些激酶或抑制剂进行体外研究。
因此,诺华和波恩团队对III型和IV型配体口袋的实验数据的调查确定了29个激酶。Rastelli团队采取的识别III型口袋的前瞻性方法发现了25个,其中很少有可以被描述为纯变构口袋的。在对这两组数据进行比较时,有两个值得注意的发现。首先,两个数据集的重叠程度不大,只有四个激酶有共同之处(EGFR T790M、ABL1、ITK和CDK2,图3中的红圈。其次,综合结果表明,所有激酶中只有不到10%的激酶可能具有III型变构口袋。然而,这可能反映了到目前为止的实验方法和感兴趣的目标,以及预测方法的口袋限制和对接确认步骤中使用的配体。理想情况下,通过多样化的筛选方法、配体集合和筛选方案,可以被变构抑制的激酶库将被大大拓宽。
其他更普遍的识别变构口袋的方法已经被报道,还有汇编相关X射线数据的数据库。其中包括激酶图谱,这是一个由FTMap在376个不同的激酶的4901个结构中确定的潜在IV型热点的数据库。尽管如此,如上所述,这些方法仍然存在两个挑战:第一,根据静态X射线结构预测异源的诱导配合机制;第二,确认一个结合口袋与目标的细胞功能活动有关。
MEK1/2和BCR-ABL1的变构抑制剂的发现史说明了在识别变构抑制剂和验证其功能活性方面的挑战和策略,值得仔细研究。
3.1 发现MEK1/2的III型抑制剂
1995年报道了色满结构1(图4),它是在一个激酶级联实验中发现的,该实验用于鉴定MAP激酶RAS/RAF/MEK/ERK信号传导途径的抑制剂。随后的实验确定MEK1/2为目标,并确定这些苗头与ATP无竞争关系。色满的细胞活性很差,但它是一个有用的参考化合物,可以进行额外的筛选工作,利用相同的激酶级联实验和细胞实验,在用化合物处理和用血小板衍生的生长因子(PDGF)刺激激酶通路后,测量磷酸化ERK(pERK)的水平。通过这一过程确定了茴香酸2,其适度的细胞活性被归因于羧酸的存在。研究了各种酸的同位素,包括羟肟酸和羟胺酯,结果是CI-1040(3)1999年报道了3在异种移植模型中的体内概念证明(POC),2000年进入临床开发。结直肠癌、非小细胞肺癌(NSCLC)、乳腺癌或胰腺癌的2期研究中,患者连续接受口服CI-1040,每次800毫克,但由于疗效有限,3的开发于2004年终止,原因是与不溶性和快速代谢有关的低全身暴露。研究小组预计到这些与3号的药理和药物特性有关的潜在挑战,并发现在两个领域的修改导致了细胞效力以及溶解性和代谢稳定性的明显改善。
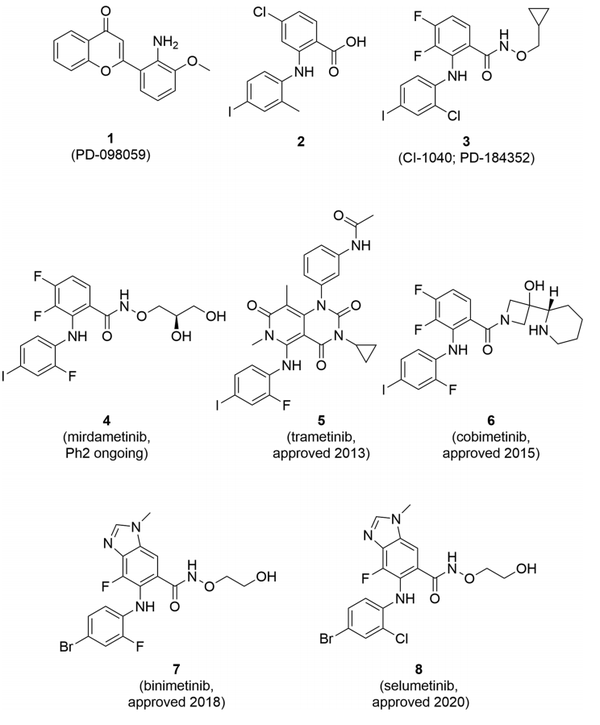
图4. III型MEK1/2抑制剂。(图片来源:J. Med. Chem.)
首先,用羟乙基羟肟酸酯类似物替换环丙基甲基基团后,细胞效力提高了10倍。其次,在苯胺环的正向位置用氟代替氯,使细胞活性提高了35倍。虽然有效力,但溶解度仍然不大,用二羟基丙基取代羟乙基酯产生了理想的效力和溶解度,并增加了代谢稳定性。(35) 这种化合物,PD-0325901,现在被称为mirdametinib (4),并仍在调查实体瘤和小儿低级别胶质瘤。这些报告激发了许多其他团队来确定MEK1/2的III型抑制剂(图4)。
早期的报告表明,3不是一个正交的结合剂;因此,研究开始解决人类MEK1和MEK2与MgATP和3的类似物的三元复合体的三维结构。这些复合体显示,抑制剂诱发了几个构象变化,导致催化不活跃的物种,并且抑制剂结合在一个与ATP位点相邻,但分开的口袋,
这种结构信息并没有影响确定曲美替尼(5;JTP-70902;GSK1120212)的筛选工作。在这种情况下,在一个高通量的支链DNA试验中筛选了16万个化合物,以确定其诱导p15INK4b mRNA表达的能力。一个最初的有希望的化合物拥有一个2,4,7-三氧吡啶并[4,3-d]嘧啶的核心,它的优化提供了5。在优化cobimetinib(6;XL518;GDC-0973)时,研究小组在MEK1:AMP-PCP复合物中应用了4的X射线共晶体结构,旨在替换羟胺酯以提高代谢稳定性。在变构口袋中利用了与Asp190的额外氢键作用。优化的结果导致了6被发现,其中3-羟基-3-[(2S)-哌啶-2-基]氮杂环丁烷酰胺是关键的结构部分。寻找和优化binimetinib(7)和selumetinib(8)相关的苗头化合物的策略尚未报道,尽管可以推断X-射线共晶体结构被应用于基于结构的方法中。
MEK抑制剂trametinib(5)、cobimetinib(6)、和binimetinib(7)的临床效用主要是与RAF抑制剂联合治疗具有BRAF V600E或V600K突变的黑色素瘤。联用的理由与对MEK1/2的变构作用机制无关,是因为对MEK的抑制会诱发BRAF通过ERK的矛盾激活。最近,III型MEK抑制剂在治疗小儿神经纤维瘤病1型方面取得了临床概念证明,导致塞鲁米替尼(8)获得批准,并正在进行成人的mirdametinib(4)的试验。
3.2 BCR-ABL1第四类抑制剂的发现
ABL001/asciminib(9)是一个IV型抑制剂的例子,相关结构见图5。诺华公司在2006年首次披露了一种用于BCR-ABL1的新型变构抑制剂GNF-2(10)。该化合物是在对50,000个化合物进行的差异性细胞毒性筛选中被优化的,该筛选是为了优先选择BCR-ABL1驱动细胞的抑制剂。最初的命题,氨基嘧啶11,很容易被认为是一种正交抑制剂,但其生化和细胞活性的其他方面与正交结合不一致。这表明,11可能是在另一个部位结合的,可能是一个调节性的肉豆蔻部位,其X射线晶体结构已于2003年发表。有了这个假设,诺华公司的团队对这个IV型口袋的功能活性进行了详尽的特征分析和验证。所采用的工具包括在肉豆蔻口袋中进行诱变以废除结合,从细胞提取物中拉出目标,以及与ATP无竞争性的动力学特征。提出了一个结合假说,随后被X射线和核磁共振结构证实。
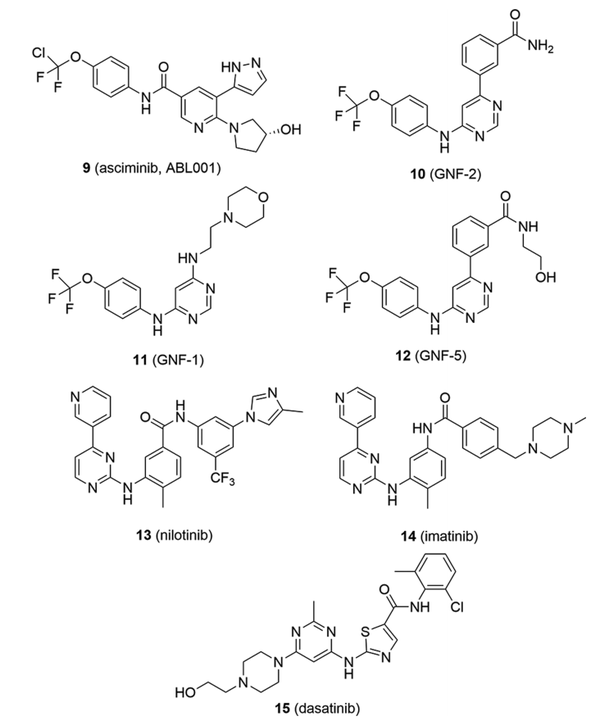
图5. BCR-ABL1 IV型和II型抑制剂。(图片来源:J. Med. Chem.)
10的优化提供了GNF-5(12),以支持小鼠体内的POC,但PK要求该团队确定一个新的起点。对500个可溶性化合物进行了核磁共振片段筛选以确定新的起点。早期苗头化合物不是功能性抑制剂,但核磁共振研究和X射线晶体结构都导致了洞察力和修改,最终产生了asciminib(9)。结构研究显示,12的活性构象是延伸的,模仿了肉豆蔻酸的构象,它的结合诱发了一个螺旋的弯曲构象,据信会导致ATP结合位点的动态或构象变化,这反过来会破坏催化机制。然而,ATP竞争性的II型配体仍然可以在12存在的情况下结合,诺华团队探索了一种双重用药方法,即9与正构抑制剂如尼罗替尼(13)共同用药。
双重用药策略的核心是这样一个假设:利用不同的靶向机制对BCR-ABL1进行双重抑制可能会通过提供增强的靶点覆盖和防止耐药性的出现来改善治疗效果。诺华团队断言,防止耐药性的出现可能取决于两种分子的耐药机制不重叠。因此,通过在含有单点突变的Ba/F3细胞中分别用9和正构抑制剂(包括尼罗替尼(13)、伊马替尼(14)和达沙替尼(15))进行48小时的增殖试验,获得了抗性谱。这些突变与肉豆蔻部位、SH3/激酶结构界面和催化部位有关。观察到不同程度的互补性,9和14具有最大的互补性,而9和13的特征有一些重叠。然后在体内对这些发现进行了探讨,因为9和13的互补性抗药性特征表明,这种组合可能会延迟抗药性,并且比单独使用任何一种药剂都有更持久的效果。事实上,在KCL-22小鼠异种移植模型中,通过同时使用9和13的治疗,达到了最大的临床前疗效。在表达BCR-ABL1T315I/H396R的Ba/F3细胞的小鼠异种移植中,9与波那提尼的组合也有类似的结果。虽然2021年底FDA最初批准9药物作为单药使用,但正在进行临床试验,研究9药物与正交抑制剂(如13、14和15)在慢性骨髓性白血病(CML)中的组合。
这两个案例研究中引人注目的是发现和开发的时间表。从首次发表变构机制到获得批准所花的时间为15至18年。MEK1/2的非正交机制首次发表于1995年,而第一个批准是在近20年后的2013年。IV型BCR-ABL1抑制剂于2006年首次在文献中报道,2021年获得批准。
接下来我们调查了文献中报道的变构激酶抑制剂的选择性和筛选方法,然后讨论如何验证其功能活性和建立变构激酶项目的流程方案。
4.1 选择性
开发变构激酶抑制剂的一个核心价值主张是,与正构部位相比,变构口袋中的残基和构象具有更大的异质性,使激酶的选择性更容易实现。虽然拉帕替尼和卡帕替尼等正构抑制剂表现出良好的选择性,但它们往往拥有以类似方式排列的共同结构特征,如氢键供体/受体(与铰链相互作用)与芳香环(模仿ATP基)相连,提高了击中其他激酶和进入日益拥挤的专利空间的机会。一般认为,激酶变构位点的同源性较低,甚至在同一激酶的异构体内也是如此,尽管文献中很难找到这方面的量化数据。然而,在一组13个ATP结合蛋白(不仅仅是激酶)中的ATP变构和正构位点的序列比对显示,相对于正生位点,变构位点的残基保护得分明显较低(0.63 vs 0.83)。此外,变构位点也可以位于不同的结构域上,为利用更大的化学多样性提供进一步的机会。例如,变构CHK1抑制剂16(图6)结合在激酶的C端结构域附近,而变构AKT抑制剂17结合在激酶和pleckstrin同源结构域之间的界面上。
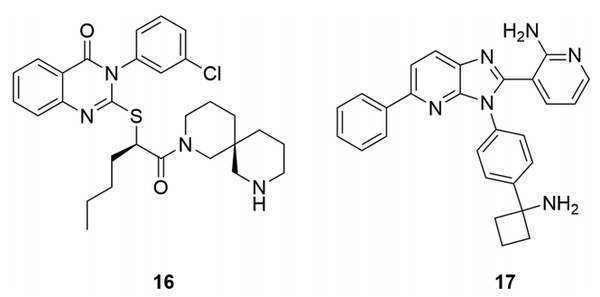
图6. 异构体抑制剂16(CHK1)和17(AKT)。(图片来源:J. Med. Chem.)
尽管变构激酶抑制剂被认为具有更高的选择性,但我们没有找到一个全面的数据集来系统地分析和证明这一类化合物的选择性。我们从文献中汇编了15种有报道的激酶选择性数据的变构抑制剂(图7和表2),所有这些抑制剂都有X射线共晶体结构,显示它们在变构位点结合,并且有生化或细胞活性,显示它们具有功能性。虽然在理想情况下,细胞数据可以用来评估一个异体抑制剂的激酶选择性,但使用一个生化检测小组来测量非目标的活性是比较常见的。在比较不同检测方法的IC50值时要谨慎,在生化检测中,ATP浓度通常为单个激酶的Km,一个化合物的细胞选择性可能与生化检测中获得的选择性不同。此外,只测量化合物破坏正交结合能力的选择性试验不应用于评估异体抑制剂的选择性,因为异体抑制剂可能不会与正交配体产生竞争。在下面的大多数情况下,每个目标只有一个化合物的选择性数据被包括在内,但我们也包括了一些结构不同的化合物,它们的目标是相同的激酶和结合部位(MEK、TrkA和AKT)。虽然存在潜在的发表偏差,因为非选择性激酶抑制剂不太可能被公开,但这些化合物中的大多数都表现出最小的非目标激酶活性。例如,TrkA抑制剂18,是一种III型化合物,具有很好的选择性。由于ATP结合部位的氨基酸残基相同,I型和II型TrkA抑制剂对TrkB和C缺乏选择性。通过在激活环和并膜结构域形成的部位结合,并利用三种异构体之间的差异,18对TrkB(180倍)和TrkC(70倍)具有选择性。它对广泛的激酶小组和Cerep脱靶小组也有选择性,在10μM时没有观察到明显的抑制作用(>40%)。另一个结构不同的TrkA抑制剂19也显示出极好的选择性。同样有趣的是,为MEK开发的几种结构不同的III型抑制剂(5、6和8)都有很好的选择性,这表明变构口袋提供了内在的选择性。
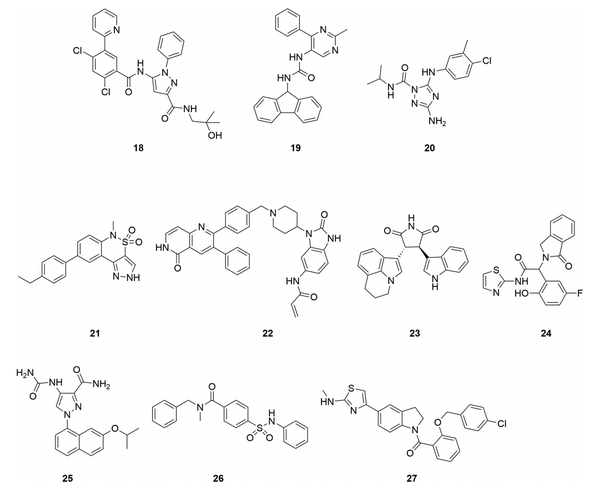
图7. 具有已公布的激酶选择性数据的变构激酶抑制剂。(图片来源:J. Med. Chem.)表2. 选定的变构抑制剂的激酶选择性
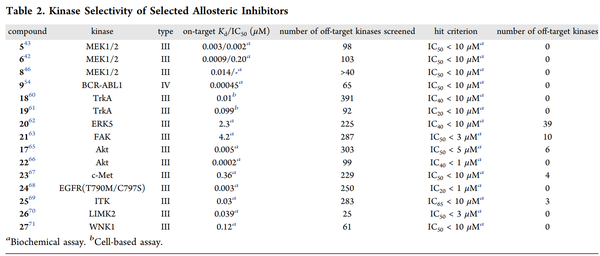
如ERK5抑制剂20和FAK抑制剂21所示,表2中的所有异体抑制剂并非都具有精致的选择性。这两个化合物对其靶点都有微摩尔效力,可能只是早期线索。与两个正交抑制剂相比,化合物20抑制的非目标仍然较少,而在21中的吡唑NH上添加一个乙基以破坏与激酶的潜在铰链结合,则大大增加了其选择性。Bajorath等人还对激酶的变构位点进行了详细的分析,并确定了潜在的交叉作用,如c-ABL和CDK2共有一个类似的结合位点,这有助于优先确定选择性分析的非目标。除了激酶之外,变构激酶抑制剂对其他类别的蛋白质如GPCRs和离子通道仍可能有脱靶活性。例如,某些基于二苯二氮卓类支架的PAK1抑制剂也抑制组胺受体H1和毒蕈碱受体M1。通过优化,这种脱靶活性被去除。
4.2 筛选方法
除了选择性之外,我们(指原作者,下同)还调查了文献中报道的所有变构激酶抑制剂的苗头生成方法。在大多数情况下,除非独立使用两种不同的方法,否则只包括发现每个激酶靶点的第一个变构抑制剂。这是因为一旦确定了 "I型"变构抑制剂及其结合位点,其他方法,如通过放射性配体置换、骨架变形和基于结构的设计进行筛选,就可以产生更多的优化线索。我们的数据集(支持信息表S2和其中的参考文献)包含17种独特的激酶,各种苗头生成方法见图8。高通量筛选(HTS)是最流行的方法,它通常使用生化或细胞试验进行。筛选中使用的库的大小跨度很大,从几千到250万。例如,发现EGFR双突变体L858R/T790M的变构抑制剂的筛选,在生化实验中检测了250万个化合物,然后用较高浓度的ATP和野生型EGFR进行反筛选,分别去除ATP位点的结合剂和非选择性抑制剂。HTS之所以占到命中率的65%,可能有其历史渊源,因为制药业经常将其作为主要的命中率寻找方法。还值得注意的是,虽然异体口袋可以提供选择性,因为相对于正交部位的序列同源性较低,但其结果是,可以被视为与正构抑制剂的铰链结合模块相同的 "优势"的化学类型较少。因此,涵盖大量化学多样性的HTS事实上可以成为寻找异体激酶的必要和理想的方法。虽然目前还没有关于DNA编码库(DEL)筛选出的变构激酶抑制剂的报道,但DEL已经成为一种重要的命题生成方法,我们预计在不久的将来会有这样的案例研究报道。
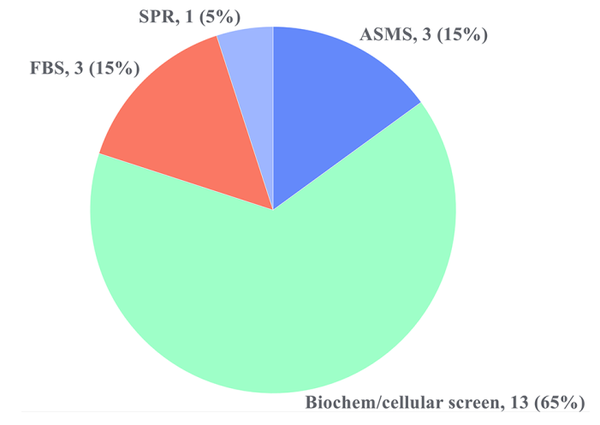
图8. 已报道的变构激酶抑制剂的苗头寻找方法。(图片来源:J. Med. Chem.)
除了HTS,生物物理方法,如表面等离子体共振(SPR)和亲和选择质谱(ASMS)也被用来发现异体抑制剂,尽管其产量可能较低。此外,虽然这些筛选可以确定与目标蛋白结合的分子,但命中的分子是否在变构位点结合或具有功能效应仍需评估。
由于文献中往往不清楚基于片段的筛选(FBS)中使用了哪些生物物理方法,我们将FBS归入命中率寻找方法的一个单独类别,通常可以使用SPR、NMR或X射线晶体学来进行。在发现变构PAK1抑制剂时,以片段为基础的筛选,然后用X射线晶体学确定了III型口袋和二苯并二氮杂卓的命中。
在筛选变构激酶抑制剂时,需要考虑到几个因素。为了发现IV型抑制剂,全长蛋白比激酶结构域更受欢迎。另外,由于构象选择或诱导拟合,异体抑制剂可能具有较慢的结合动力学;因此,较长的测定时间可能有助于发现此类化合物。此外,在结合试验中,可使用已知的正则结合剂来阻断ATP位点,从而过滤掉正构结合剂。另外,还可以利用反筛选或计算机模拟过滤,从苗头中去除正构结合剂。
率先发现这些不同激酶的III型和IV型抑制剂的团队所报告的经验表明,即使有良好的筛选策略和大量的化合物收集,验证一个新的口袋并确认变构结合转化为感兴趣的功能活性可能是一个漫长的过程。虽然基于细胞的筛选可能是成功的,但确认结合模式的输出去卷积可能是耗时的,发现BCR-ABL1 IV型口袋就是证明。如果选择HTS方法,那么变构口袋本身的属性可能会增加评估筛选结果的复杂性,特别是在识别IV型口袋时。首先,波恩小组对激酶结晶集的分析表明,虽然可以发现大量的口袋,但其中许多是浅层的。因此,在进一步优化之前,筛选出的结果可能亲和力不大,而且没有功能活性。其次,变构口袋已被证明比正构位点更亲脂,因此,所找到的基因可能具有较差的物理化学特性,必须在随后的优化中加以解决。由于变构位点的残基性质更加多样化,相应配体的结构特征也是多种多样的,由于缺乏特殊的异体抑制剂化学类型,必须对命中的化合物进行仔细的筛选。
在这种情况下,线索生成流程方案必须明确地确认变构结合,以及解决潜在的温和效力和差的物理化学特性,因为它们被优化为功能活性。详细介绍筛选异体抑制剂的许多方法超出了本综述的范围,但筛选方法应允许识别并从命题组中去除正交结合物,并进行第二层评估以确认结合亲和力并确定结合部位(图9)。根据活性物质的亲和力,可以通过SPR、ASMS或差示扫描荧光法(DSF)来评估结合情况。生物物理学方法,如X射线晶体学、冷冻电镜和核磁共振,可用于确定配体结合部位。在这个阶段,分析检测可以包括通过动态光散射(DLS)评估化合物的聚集倾向或对酶浓度变化的敏感性。在有代表性的活性物质存在的情况下,对酶的动力学进行分析可以达到两个目的。首先是了解配体是ATP竞争性的还是非竞争性的。III型配体主要是ATP非竞争性的,尽管如上所述也有例外。第二是了解配体的导通率,因为异体结合可以对应缓慢的结合现象,这就表明需要调整检测条件,如延长检测时间,以达到真正的平衡。随着化合物系列的成熟和亲和力的优化,通过生物物理方法进行的结合确认可以从关键路径转移到抽查,并且可以在测试漏斗中加入功能性生化检测。在向 "苗头"到"先导"或 "先导优化"的过渡中,对功能活性的评估将以细胞为基础,采用相关的药效学终点,如下游激酶的激活,而不是抗增殖活性这样的表型。
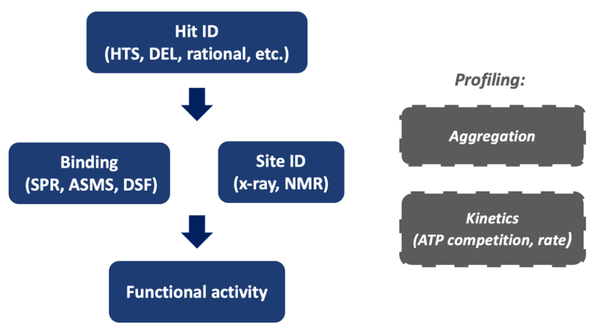
图9. 建议用于表征和优化变构抑制剂的流程方案的组成部分。(图片来源:J. Med. Chem.)
相对于正交激酶项目的流程方案,其中两个功能检测通常就足够了(即生化和基于细胞的检测),确认变构配体的结合和确定其位置不能被视为理所当然,变构抑制剂的验证和优化需要评估结合和功能活性。通过仔细的生物物理表征为项目打下基础,可以减少或避免后期的返工。
在过去的十年中,FDA批准了变构(III型和IV型)激酶抑制剂,验证了这些结合模式在临床上是可行的。目前,相对于整个激酶组而言,少数激酶被报道有III型或IV型口袋,可以理解的是,调查的重点是已经建立体内临床前或临床POC的激酶。MEK1/2 III型抑制剂的历史表明,如果有经过验证的工具化合物,这些努力会变得更容易。要在更多的激酶中找到III型或IV型口袋,需要利用过去20多年来获得的经验。面临的挑战包括:这些口袋可能是浅层的和瞬时的,从而导致通过实验和计算方法识别它们的复杂性。因此,确认结合导致理想的功能后果可能是一个劳动密集型和耗时的经验。
令人鼓舞的是,在激酶组的每一个亚家族中都有变构口袋的报道,而且计算方法表明,更多的激酶含有III型口袋,这些口袋仍有待配位。异体抑制剂的选择性和耐受性的提高,为继续探索激酶组和识别这些蛋白质提供了动力。最近几个组织关于PI3Kα突变体的变构抑制剂的报告显示,人们对这个领域的兴趣仍然很高。asciminib的临床前疗效与同一激酶的正交抑制剂结合起来具有很强的活性,再加上明显减少的突变趋势,进一步支持在其他激酶中寻找类似的机会。有了广泛的配体寻找策略,如酶法和基于细胞的HTS,以及基于片段和DEL的筛选,发现工具箱已经准备好迎接挑战。
Pan Y, Mader M M. Principles of Kinase Allosteric Inhibition and Pocket Validation[J]. Journal of Medicinal Chemistry, 2022.